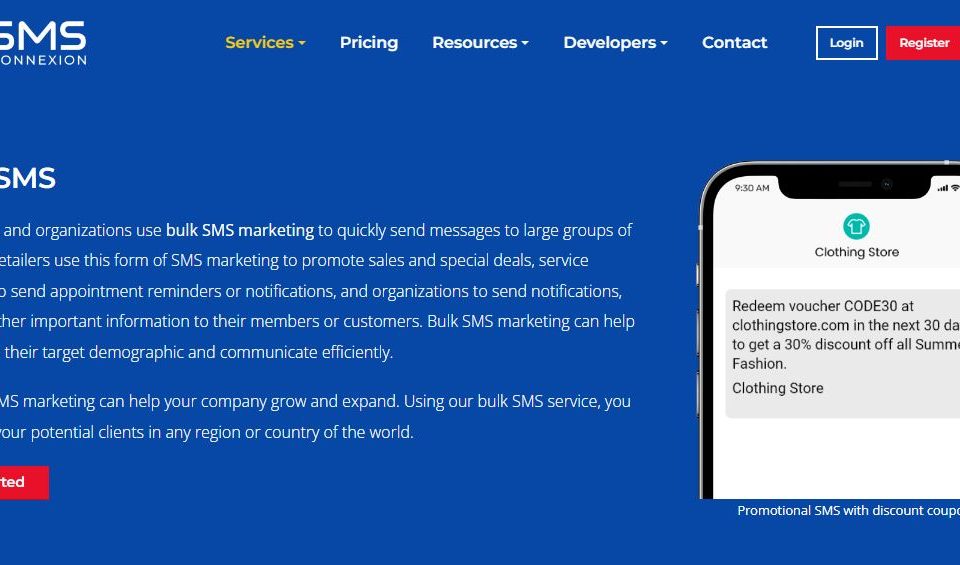
The Benefits of Bulk SMS For Businesses
There are a variety of benefits to sending bulk sms to your customers. Some of these include improving your sales and brand awareness. Others include
Finance
There are a variety of benefits to sending bulk sms to your customers. Some of these include improving your sales and brand awareness. Others include

If you have a need for a personal loan, then you need to make sure you are aware of the various factors that will affect
While dating a younger woman, men should remember to be patient and considerate. She may be younger than you, but her inner soul is still
Many men wonder how to find a younger woman. Some assume that it will be easier to meet a girl their own age, but this

Many people think of leisure travel when they think of tropical destinations. Bali, Malaysia, Mexico, Puerto Rico, the Bahamas, Turks, and Caicos Islands, and Thailand

A Travel Guide is a licensed tour guide who takes groups of tourists to various destinations. These guides plan complete itineraries that include recreational activities

Technology has various meanings. It may mean something in the form of a sci-fi movie, a machine, an electrical device, or a simple technique. It

Communication Technology has revolutionized our society. In the past, social interactions were largely governed by political factors. With modern communication technologies, social interactions are governed

If you’re thinking of buying a commercial property, you should consider the location and resale value of the neighborhood. After all, you will be 100%

Choosing a real estate agent should not be based solely on price. You should look for a professional who can provide you with valuable advice,

If you want to increase the value of your home, you must consider doing some home improvements. The living room, dining room, and bedroom all

When it comes to landscaping, one of the first things to think about is how the landscape will affect the overall atmosphere of the house.

When choosing what to gift, there are four basic types of gifts: Experience, Knowledge, Helping, and Prophecy. Each one has its own value, and each

Choosing gifts for kids can be tricky, but don’t worry! Girls love getting new toys and electronics, and they love to follow their favorite celebrities.

If you have been considering becoming a part of the corporate finance world, this article will give you some helpful advice. This field is vast

The current state of global public finance is challenging. Governments are grappling with profound fiscal imbalances and systemic financial failures. Under intense pressure, public finance

A general description of finance is outlined in the following paragraphs. In a nutshell, finance includes four main areas: Personal, Public, Corporate, and Accounting. Each

If you’re dating someone, you have to keep a few rules in mind. For example, you should never engage in sexual intimacy until you are

Identifying red pill sexual strategies and how to avoid them are vital to a healthy relationship. Learn the secrets of successful dating and avoid committing

While dating is a type of fun, you can also date many different people at a time. Dating is all about having fun and developing

Whether it’s a birthday, Christmas or Mother’s Day it is a time to celebrate the most important women in your life. This year, treat your

If you are just starting out, you may be wondering how to begin investing in stocks and shares. This article can help you understand the